

Under de senaste decennierna av min karriär har jag ägnat otaliga timmar åt att arbeta för att skydda amerikaner genom att undersöka drogernas säkerhet. Min utbildning och karriär har tagit mig genom ett halvdussin universitet, Big Pharma, och vid FDA under tre presidentadministrationer. Läkemedelssäkerhet tar hänsyn till varför en individ kan ta en läkemedelsprodukt och ha noll biverkningar, medan en annan individ kan ta samma produkt men har biverkningar upp till och inklusive permanent funktionsnedsättning eller dödsfall. Som standard tar studier av läkemedelssäkerhet också hänsyn till icke-kliniska aspekter av tillverkning och läkemedelskvalitet.
Eftersom läkemedelskvalitet är en viktig faktor för att bedöma läkemedelssäkerhet, ledde min vandring för att skydda amerikaner till att jag konceptualiserade och grundade världens första "analytiskt apotek” uppdrag med att vetenskapligt verifiera farmaceutiska produkter från platser som Indien och Kina innan de distribueras till patienter. Tyvärr ledde jakten på stort över etik och skydd av patienter till att företagets ekonomiska ledning engagerade sig omfattande FDA-överträdelser och anklagas av domare för att göra falska vetenskapliga påståenden (som alla av misstag inträffade efter min utgång).
Utan extern bekräftelse av läkemedelskvalitet är amerikaner helt beroende av FDA och tillverkare för att bedöma och bekräfta produktens renhet. Läkemedelssäkerhet har visat sig vara ett anmärkningsvärt problem när det gäller Covid mRNA-injektioner. Tyvärr, om någon ville göra sin egen analys på mRNA-injektioner, de inte har en lämpligt detaljerad ingredienslista att jämföra den med, eller ens tillgång till den etablerade regulatoriska metoden för hur man korrekt testar den för renhet.
Det är för att tillverkare och FDA överväger alla ingredienser i dessa mRNA-injektioner, inklusive sekvensen av mRNA plus egenskaper för lipidnanopartiklar (LNP), inklusive halveringstid, LNP-strukturer, ytmodifiering(er), antal/typ(er) av LNP per dos och fästpunkter på mRNA-strängen, att vara ospecificerad eller "affärshemlighet".
Ovanpå det anser FDA dessutom att metoder om hur man testar mRNA-injektioner för renhet en affärshemlighet också.
Bipartisan stöd och hundratals miljarder skattebetalare, men INGEN insyn?
Covid-mRNA-sekretess existerar även om både Trump- och Biden-administrationerna hade föreslagit full transparens med mRNA-injektioner till den grad att Covid-mRNA-immateriella rättigheter hävdes. Trots det tillåter både FDA och tillverkarna patenten, inklusive grundläggande data om dessa skott, som en affärshemlighet. De gör det trots att alla Covid-vaccintillverkare har fått hundratals miljoner skattebetalare enligt Forbes/Statista publikationer.
Att studera drogsäkerhetsepidemiologi är tillräckligt svårt. Utan verifierbar produktrenhet/konsistens är en fullständig säkerhetsutvärdering omöjlig.
Full transparens för alla ingredienser och kvalitetskontrollåtgärder är viktiga inte bara för att de finansierades kraftigt av skattebetalarna med hundratals miljoner dollar, utan för att en rad frågor har uppstått om säkerheten och effekten av Covid mRNA-injektioner.
Förutom att vara exceptionellt komplexa, påskyndades deras godkännande av tillsynsmyndigheter efter mindre än ett år. De flesta läkemedel och vacciner tar vanligtvis runt tio år att fullständigt testa för säkerhet/effektivitet och granska och godkänna. Förutom att ingredienserna är helt nya, mycket komplexa och de första i sitt slag som administreras i massiv skala, utvecklas bl.a. långtidsutvärderingar av klinisk säkerhet/toxicitet och epidemiologiska granskningar påskyndades och sannolikt inte helt klarlagda före utgivningen.
FDA-ingrediensverifiering, transparens och "sanning" har prejudikat som går tillbaka till 1800-talet:
Den analytiska verifieringen och insynen av ingredienser eller "sanningen i märkning" där innehållet i flaskan är Obligatorisk för att matcha de listade ingredienserna före etableringen av FDA, tillbaka till 1862. Dagens FDA föddes faktiskt ur det som började som en enda "Department of Chemistry"-anställd anställd vid US Department of Agriculture.
Förfalskning, (förändrade eller giftiga ingredienser) felmärkning (innehåller en falsk etikett eller är på annat sätt vilseledande, eller innehåller felaktiga medicinska påståenden), eller felmärkning (innehåller ingrediens(er) är inte listade på en produktetikett) har alla haft långa, fula historia i Amerika. Man trodde att eländet hade nått sin topp i början till mitten av 19-talet – eller åtminstone då det blev identifierbart – eftersom det först 1862 hade tekniska processer utvecklats för att analysera och upptäcka ingrediensbedrägerier. Dessförinnan skulle så kallade "resande medicinmän" som kallar sig "läkare" (alltid med tvivelaktiga eller obefintliga meriter) slänga flaskor med "bota allt"-produkter, vars ingrediensetiketter endast skulle lista skumt eller ofarligt innehåll som t.ex. "vitaminer""örtextrakt,"Eller"orm olja” – eller har ofta ingen ingredienslista alls.
På den tiden, många fromma, puritanska New Englanders, som av religiösa skäl skulle aldrig beröra alkohol, skulle köpa dessa lösningar från dessa tuffa hucksters och omedvetet luras till att konsumera lösningar som inte bara innehöll alkohol, utan narkotika som opium och/eller kokain. Under sken av att förbättra ett absurt brett ymnighetshorn av åkommor, utvecklade patienter istället ett straffberoende och/eller fick på annat sätt sin hälsa negativt påverkad av dessa tidiga "droghandlare".
När problemet växte började den federala regeringen ta hänsyn. Så småningom, den Pure Food and Drug Act antogs 1906 och ledde till skapandet av Food and Drug Administration (FDA).
[FDA hade en formande skyldighet att se till att droger har sanningsenliga märkningsuppgifter och uppfyller vissa standarder för renhet och styrka.
Kom ihåg att nästan 120-åring sanningsenliga märkningskrav och "renhets"-delen av Pure Food and Drug Act från 1906 när du läser om mRNA-verifieringstestning och ingredienstransparens.]
Vilka "sanningsfulla" och "rena" tester för ingrediensverifiering förekommer för FDA-reglerade produkter?
Redan 2021 valde FDA att börja övervaka USA:s läkemedelskvalitet via en fjärrinsamling of inskickad inlämning av prover för droger som ersättning för inspektioner av levande anläggningar på grund av Covid-pandemin. Var det lagligt? Kan det någonsin anses vara vetenskapligt lämpligt? Idag, trots att pandemin har upphört, är den enda officiella testning av läkemedelsfrisättning som för närvarande utförs på vilken som helst Covid mRNA läkemedel visas till fortfarande göras av FDA via en tillverkare tillhandahållen, "inskickad" prov enligt a skärmdump av den aktuella FDA-webbplatsen. Uppenbarligen är en "inskickad" provtagningsmetod mycket annorlunda och potentiellt mindre tillförlitlig än att direkt samla in prover via en direkt, personlig insamlingsmetod. Trots det hävdar FDA att de har "den högsta standarden över hela världen för provtagning och testning. "
Dessutom föreslår FDA att ytterligare utveckla sin "inskickade" fjärrtestpolicy med en nyligen föreslagna vägledningsdokument.
Även om det bara existerar som ett "utkast" till FDA-dokument, visar officiella FDA-webbplatser det utskicket av prover verkar redan ha implementerats sedan åtminstone januari 2021. FDA verkar hävda resultaten av dessa insända tester som deras oberoende verifiering.
Dessutom längst ner på första sidan av FDA-utkastet dokumentet föreslår utvidgning av "fjärrtestning." Den listar för närvarande varje FDA:s produktreglerande avdelning vid FDA, vilket antyder att det är ett byråövergripande policyförslag.
Den kompletta listan inkluderar:
- Office of Regulatory Affairs
- Office of Food Policy and Response
- Kontoret för kombinationsprodukter
- Centrum för biologisk utvärdering och forskning
- Centrum för läkemedelsutvärdering och forskning
- Centrum för enheter och radiologisk hälsa
- Centrum för livsmedelssäkerhet och tillämpad nutrition
- Centrum för tobaksvaror
- Centrum för veterinärmedicin
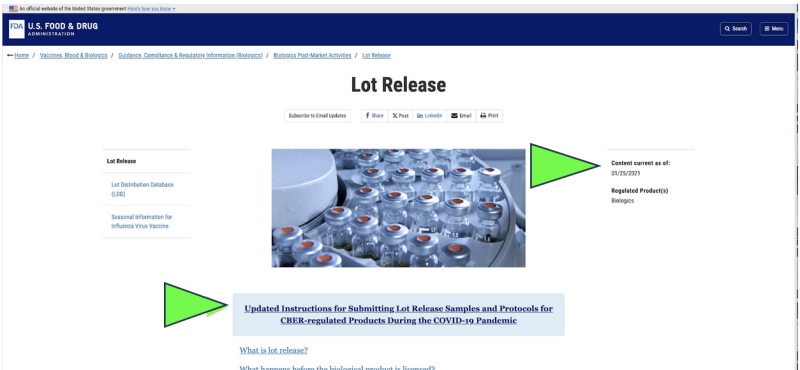
Är "inskickad" kvalitetskontrollprovtagning av FDA lämpligt? Vad händer om statens hälsoavdelnings restauranginspektioner speglade FDA:s policy?
Denna "in-in-"-provtagningsmetod är lika absurd, till exempel för att en delstats hälsoavdelning övervakar restauranger genom att be dem att regelbundet "posta in" olika föremål från deras meny till en testanläggning så att hälsoavdelningarna kan testa för potentiell mat -född kontaminering och/eller ber restauranger att lova att själva testa menyalternativ. Tänk om den restaurangen låg i Kina? Tänk om den restaurangen låg i Indien? Eller något annat land som är välkänt för att ha en urusel historia av bedrägerier och kvalitetskontroll problem?
Den metoden skulle vara oacceptabel för både restauranger och läkemedelsföretag, av skäl som inkluderar det uppenbara: tillverkare kan skicka in de prover de föredrar – inte nödvändigtvis representativa batchprover. Det är uppenbarligen inte samma sak som att FDA-inspektörer skaffar prover under oanmälda inspektioner av hela anläggningen.
Under restauranganalogin skulle naturligtvis alla restauranger göra det lämna in "A" betygsprover som inte nödvändigtvis skulle vara representativt för vad konsumenterna får.

Kvalitetskontroll: Vad är farmaceutisk "frisättningstestning" och varför är det viktigt?
Idag övervakar FDA kvaliteten och innehållet i $2.7 biljoner värdet av produkten årligen, men verkar undertrycka bedömningar och resultat av kritisk ingrediensverifiering. FDA är tänkt att skydda amerikaner genom att utföra omfattande analytisk testning som en kontrollsumma för att säkerställa ingrediensnoggrannhet. Resultaten av det borde vara transparenta för skattebetalare som finansierar FDA:s 6.6 miljarder dollar budget. Den vetenskapliga verifieringen kallas för farmaceutisk "släpptestning.” Frisättningstestning är en teknisk term som hänvisar till en process som involverar en mängd olika instrumentella analyser som används för att omfattande testprodukter för renhet, koncentration, konsistens, identitet och föroreningar av alla slag.
Hela FDA föddes från den där "Department of Chemistry"-anställd från 1862 och behovet av transparens och verifiering av ingredienser. Idag har den anställde förökat sig till en hela FDA-avdelningen med 1,300 XNUMX forskare och supportpersonal förmodat tillägnad ingrediensverifiering via testning av läkemedelsfrisättning. FDA:s Office of Pharmaceutical Quality (OPQ) är tänkt att se till att läkemedel exakt matchar innehållet i de listade ingredienserna, utan kvalitet/orenhet (kvalitativ) eller innehållsvariation (kvalitativ). Reglerna som kräver det är mycket specifika och detaljerade i 21 CFR § 201.10.
Hur FDA verifierar mRNA-injektioner för kvalitetskontroll:
Kvalitetskontrollresultaten från tester från mRNA-injektioner var särskilt kritiska eftersom de är stora, komplexa och gjordes snabbt. Medan skattebetalarna är beroende av FDA för att verifiera mRNA-injektionskvaliteten och dela resultaten, menar FDA verkar skyldig att skydda tillverkarnas ingredienser på bekostnad av även den mest grundläggande transparensen gällande mRNA Covid-produkter. Medan FDA tycks samla in prover, är deras "mail-in"-metod i grunden felaktig. Dessutom delar inte FDA resultaten av dessa tester någonstans där jag kan hitta dem.
Med andra ord: under pandemin när helt nya, brett implementerade mRNA-shots sattes på amerikaner i "varphastighet" och när Amerika förlitade sig mest på FDA:s kvalitets-/regleringsplikter, accepterade FDA självinlämnade "postade- i kvalitetskontrolltestning och/eller resultat. Tänkte inte FDA på det mRNA-tillverkare medgav att de "kämpar[d]" för att svara på tillverkningen och "kryddade" för att hänga med med tillverkningsprocesser? Tillverkare av mRNA-ingredienser uppgav vidare att ansträngningarna att möta behoven var "utan motstycke."
Uttalanden som detta ger inte konsumenternas förtroende för kvalitet och är illustrativa för en enorm uppskalning av dessa komplexa produkter som borde motivera särskilt vaksam och in-person FDA granskning av anläggningar och tillverkade produkter, pandemi eller inte. En tillverkare av mRNA-ingredienser, till exempel, uppgav att de plötsligt ökade sin produktion med 50 faldig.
Mitt i den nya teknologin som drevs igenom i "varphastighet" var ingen av de 1,300 XNUMX OPQ-forskarna vid FDA som krävde liveinspektioner, eller åtminstone erbjöd sig att göra något annat än att be om potentiellt tvivelaktiga "inskickade" prover för provning?
Den uppenbara frågan är: varför samlade inte FDA in prover direkt? Även med pandemin på plats kunde FDA ha inspekterat anläggningar som bär hazmat-dräkter eller – eller på mycket minst – valde att samla in prover från apotek, sjukhus eller på distributörslager.
Dold metod för att testa mRNA-injektionsingredienser:
Utöver frånvaron av testresultat och tvivelaktiga "inskickade" provtagningsresultat – är FDA dessutom döljer sin validerade metodik som hindrar andra från att utföra sina egna, oberoende analyser av kvaliteten/renheten hos mRNA-injektioner.
Att självständigt analysera droger för renhet och potentiell kontaminering jämfört med ingredienslistan är något jag hade försökt göra själv när jag konceptualiserade världens första analytiskt apotek. Men eftersom mRNA-skott är en ny teknik med en mindre än helt genomskinlig ingredienslista, är testmetoden man skulle behöva använda inte enkel som den skulle vara för andra småmolekylära läkemedel. Alla som försöker slå upp lagring, stabilitet, specificitet, kemi, känslighet eller till och med grundläggande metodik för att testa validering och/eller resultat blockeras via en FDA-rapport som innehåller löjligt invasiva redaktioner, vilket ger till och med den mest grundläggande vetenskapliga förståelsen av hur man potentiellt kan utvärdera resultat eller genomföra tester omöjligt.
Som ett gripande visuellt exempel är en enda redigerad sida i en längre FDA regulatorisk sammanfattning (visas nedan) en del av en 127-sida dokument (endast 63 sidor har delats och av dessa 63 sidor har cirka 50 % redigerats) om hur man utvärderar renheten, koncentrationen och andra analytiska mått på mRNA-injektioner.
De FDA (b)(4) redaktioner specificerade detaljerade redaktioner som används för att "skydda affärshemligheter och konfidentiell kommersiell eller finansiell information.” Men är det verkligen lämpligt att märka det som "kommersiellt" om forskningen/utvecklingen/produkten finansierats med hundratals miljoner skattebetalare?

Utan en lista över ingredienser eller testmetod är det omöjligt för någon annan utanför FDA eller tillverkare att veta exakt hur man kontrollerar produkten förvanskning (förändrade eller giftiga ingredienser) eller felmärkning (eftersom en fullständig lista över ingredienser inklusive nukleotidsekvensen och lipid nanopartikelkonfigurationer är särskilt vaga på produktetiketten).
Bristen på metodik är särskilt besvärlig eftersom nya, preliminära data med hjälp av oberoende metodik har visat bevis för DNA-kontamination i mRNA Covid-injektioner.
Så om en utomstående individ påstod sig ha testat och hittat en förorening i mRNA-sprutor och frågade FDA eller tillverkarna om dess svar, skulle de mötas av något svar som säger något i stil med:
- Du använde inte validerad/lämplig testmetod för att komma till dina slutsatser och därför är dina analyser ogiltiga.
Till det skulle det oberoende laboratoriet försöka begära testmetoden från FDA-godkänd dokumentation (dvs. det fullständiga dokumentet som innehåller Figur 5) genom att fråga: "Okej, jag skulle vilja testa det med din godkända metodik; kan du berätta vad det är?"
- FDA eller tillverkaren skulle svara något i stil med: "Vad vi är villiga att avslöja om den metod som används som inte är konfidentiell kan hittas online eller via en FDA FOIA-förfrågan” …där de skulle mötas följande kraftigt redigerade dokument, där allt som är avlägset meningsfullt täcks av (b)(4) redigeringar.
Att läsa mellan raderna: Det är uppenbart att både tillverkare och amerikanska FDA inte vill att någon annan än de själva ska känna till de fullständiga ingredienserna i eller till och med testa mRNA-injektioner för renhet och konsistens.
Enligt FDA-tjänstemän: Pharmaceutical Manufacturing är I hög grad Felbenägen:
Många saker kan – och gör – gå fel under den farmaceutiska tillverkningsprocessen. Utöver potentiella inkonsekvenser med mRNA/LNP-injektioner, implicerar kvalitativa och kvantitativa frågor varje FDA-reglerad läkemedelsprodukt. Till och med parlamentet och senaten har formellt erkänt rapporter om FDA:s misslyckande med att säkra USA:s läkemedelsförsörjningskedja. Majoriteten av Amerikas läkemedel konsument-slutanvändarproduktproduceras utomlands i länder som Indien och Kina, och andra lågarbetskostnadsländer är inte väl ansedd för höga nivåer av kvalitetskontroll. Det federala registret är full av rapporter om kränkningar vid indiska och kinesiska tillverkningsanläggningar.
Certifierar FDA också dessa växter – inklusive de med lång historia av överträdelser – via ett "mail-in"-system till FDA? Upprörande nog är svaret på frågan något som skulle göra alla som berörs av läkemedelskvalitet väldigt obekväma.
Medan a Sex Sigma precisionsnivå har länge varit målet för kvalitet och säkerhet inom bil-, dator-, mobiltelefon- och annan högteknologisk tillverkning, det verkar mest ha förbisetts när det kommer till läkemedelstillverkning.
FDA-tjänstemän har publicerat data som uppskattar en oprecision på 2-3σ (sigma) vid läkemedelstillverkning. En 2σ kvalitet motsvarar 308,537 1,000,000 defekter per XNUMX XNUMX XNUMX tillfällen. (Det finns sannolikt mycket mer än 1,000,000 XNUMX XNUMX möjligheter till fel när det gäller läkemedelstillverkning.) FDA är medveten om detta på de högsta nivåerna av ledarskap; faktiskt strömmen FDA:s chef för Office of Pharmaceutical Quality, Michael Kopcha skrev och publicerade till och med ovanstående Six Sigma-beräkning och beklagade den oprecisa karaktären hos läkemedelstillverkning tillbaka i 2017.
Fellatituden för mRNA-produkter och/eller deras LNP:er kan vara jämn mindre exakta än 2-3σ, (ju lägre σ, desto mer felaktig är en produkt) eftersom de inkluderar nukleotidmaterial och nya LNP, vilket gör dem avsevärt mer komplexa än småmolekylära läkemedel - trots att de utvecklas, tillverkas och släpps på " warphastighet."
Med till och med FDA och dess tjänstemän erkänner en inneboende tillverkningsfel, varför i sportens vida värld uppfyller inte FDA sitt säkerhetsuppdrag genom att offentligt dela sin releasetestning av mRNA-teknik med den amerikanska allmänheten som finansierar dem?
Före 1862 igen? Är mRNA-skott de enda drogerna som amerikaner inte har Komplett Ingrediensinformation?
Bristen på klarhet i antalet sekvenser av mRNA-skott och annan kritisk information står i direkt kontrast till ett annat FDA-godkänt RNA-baserat läkemedel – patisiran (Onpattro®). Onpattro tillhandahåller transparent sekvens, molekylvikt och milligramstyrka för sina produkter inom officiella FDA förpackningsmärkning som illustreras i ett utdrag nedan:


Brist på Covid mRNA Dosspecificitet: 0.3 ml (eller 0.5 ml) av vad?
För närvarande har vi fortfarande ingen grundläggande ingrediensinformation om någon Covid mRNA-injektion. Apotekare vet bara att ge en specifik volym av vätska, och gjorde det till synes utan tvekan. Normalt bör den officiella FDA-förpackningens märkning specificera de faktiska ingredienserna i den volymen, men inte för Covid-mRNA-etiketter: de anger helt enkelt 0.3 ml (eller 0.5 ml) som "Doseringsform och styrka."
Dessutom, som alla gymnasieelever kan berätta för dig, är 0.3/0.5 ml en volym, inte en hållfasthet. Vi känner inte till några kvantitativa detaljer om vad som finns i den 0.3/0.5 ml som: Hur många LNP-partiklar? Vilken storlek/morfologi för dessa LNP? Hur många mRNA-sekvenser i den volymen?




Är detta vad som är tillräckligt transparent eller "sanningsfull märkning" av FDA?
Ovanstående klipp-och-klistra-utdrag från bipacksedeln är all information som tillverkarna delar med konsumenterna angående dosen – som är beklagligt otillräcklig jämfört med alla andra FDA-etiketter – eller någon som är nyfiken på att veta något utöver hur mycket vätska att injicera och koncentrationen på 30 eller 100 mcg av en ospecificerad mRNA-sekvens.
Den anmärkningsvärda oprecisionen i denna etikett som tillåts av FDA verkar komma i konflikt med dess nästan 120 år gamla etikett specifikt: "kräver att mat och droger bär sanningsenliga märkningsförklaringar och uppfyller vissa standarder för renhet och styrka. "
Är detta vad som passerar som en "sanningsfull" lista över ingredienser av FDA? (Ser 21CFR §352och 21 CFR §201.10 angående "deklaration av ingredienser" och "felmärkta läkemedel och anordningar.")
Frågan är: listar okända eller ospecifika ingredienser som ingen utom tillverkaren kan tyda verkligen uppfyller andemeningen eller lagkraven för "märkning?" Är den etiketten det som anses vara "sanningsfullt" av USA:s FDA? Vars sida är FDA på ändå; tillverkare eller konsumenter?
Förutom att det inte är direkt specificerat kan det exakta antalet LNP- eller mRNA-strängar i en 30 eller 100 mcg injektion inte ens extrapoleras stökiometriskt eller på grundval av Avogadros nummer, eftersom mRNA-sekvensen, molekylvikten och/eller LNP-komponenten/-konfigurationerna inte tillhandahålls någonstans inom den officiella FDA-märkningen.
Hur kan någon veta om antalet mRNA-strängar för att koda spikproteinet för Covid är proportionell mot belastningen av Covid-ympning som man skulle få från en infektion som förvärvats i samhället? Svar: de kan inte.
Är Covid mRNA-injektioner Lämpligt märkt/felmärkt?
21 CFR 211.125 anger "Strikt kontroll ska utövas över märkning som utfärdats för användning vid märkning av läkemedel,” men det verkar som att FDA var så slapp med sin godkända märkning av Covid mRNA-injektioner trots att alla andra droger – inklusive mRNA-baserad Onpattro – anger den informationen. Historiskt sett är FDA-reglerande beslut (som vilken information som ska inkluderas i produktmärkning) baserade på prioritet, och Covid mRNA-skott var en uppenbar avvikelse från FDA:s historiska och juridiska företräde. Den anmärkningsvärda frånvaron av data och brist på klarhet går liksom tillbaka till tiden Morleys lever och njure i slutet av 1800-talet. Skillnaden är: då fanns inte FDA, men idag finns det en FDA med ~20,000 XNUMX anställda, av vilka åtminstone några skenbart trodde att denna etikett var transparent och "sanningsfull".
Att ange en okänd/otydlig/obskyr ingrediens som ingen någonsin kunde avgöra exakt är inte vad lagstiftare från 1906 Pure Food and Drug Act avsåg när de specificerade FDA-reglerna om "sanningsfull märkning." Separat från det: det faktum att doserna fördubblas per volym från olika tillverkare (30 mcg/0.3 ml vs 100 mcg/0.5 ml) betyder att dessa mRNA-sekvenser verkar vara väldigt olika i nukleotidlängd och i sin tur skulle ha fler och olika LNP plus bilagor. Betyder det att mRNA-sekvenser som används för att transkribera spikproteinet är ungefär dubbelt så stora (10mcg/0.1mL mot 20mcg/0.1mL) jämfört med olika tillverkare, eller är det något annat som bidrar till skillnaden i nukleotidlängd?
För lekmannen som fortfarande läser fram till denna punkt (kudos, förresten): Bristen på detaljerad märkningsinformation kan vara som att allmänt annonsera ett hus till salu, att säga att det är gjort av trä och tegel, på en cementplatta – men inte att visa några bilder av huset, (t.ex. sekvens) och inte delar dess kvadratmeter (t.ex. molekylvikt). I vilket fall som helst är bristen på information otillräcklig och en avvikelse från traditionella standarder.
Alla andra FDA-godkända läkemedel – inklusive andra mRNA-läkemedel – innehåller fullständiga ingrediensbeskrivningar på sina produkter, inklusive en strukturell representation och molekylvikt av sin produkt så att folk vet exakt vad de får.
Det är sant: Slå upp vilken drog du än kan tänka dig i Drugs.com databas och notera hur alla etiketter ger struktur och/eller molekylvikt. Bevis på att Covid-mRNA-skott är en iögonfallande undantag från FDA:s historiska godkännandepraxis och regeln om "sanna etikett".
2023 danska studiedetaljer Signifikant klinisk variation mellan batcher av mRNA Covid-19 mRNA-injektioner:
Att inte ha någon insyn i ens potentiellt ogiltiga "inskickade" testvalidering verkar ha gett tillverkarna en passning på en annan kritiskt viktig del av vad FDA övervakar: potentiella kliniska manifestationer på parti-/batchvariationer av mRNA-skott. En retrospektiv Dansk säkerhetsstudie publicerad tidigare 2023, detaljerade ett mycket avvikande mönster av biverkningsrapporter från Pfizer-BioNTech BNT162b2 mRNA-injektioner som korrelerade med det danska DKMA:s biverkningsrapporteringssystem.
I linjediagrammet som följer representerar olika färgade prickar olika partier av Pfizer-BioNTechs mRNA-injektioner. Den delade upp partier i tre olika kategorier; hög-låg- till (nästan) frånvarande antal rapporterade biverkningsgrupper (blå, grön respektive gul plot).
Med andra ord: Förmodat "likvärdiga" produkter från samma tillverkare verkar ha väldigt olika förekomster av biverkningar, per parti, där var och en av dessa partier representerar hundratusentals mRNA-injektioner.
När motsvarande linjära regressionslinjer lades till, uppstod ett speciellt mönster:
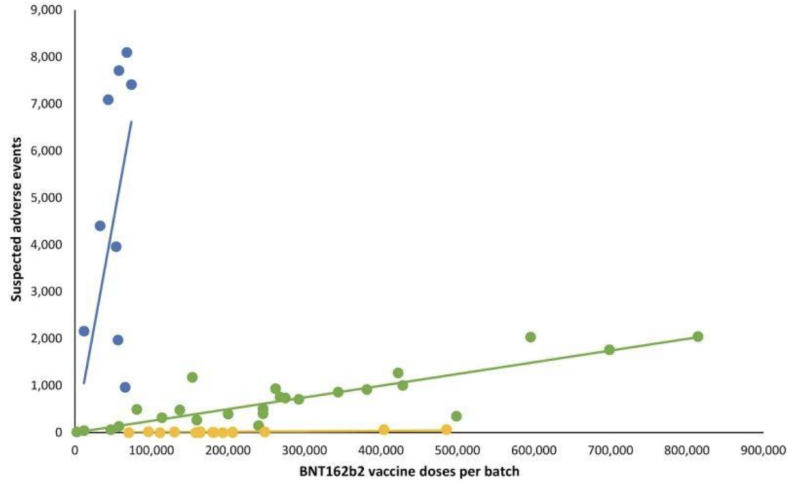
Viktiga frågor om de anmärkningsvärda oönskade händelsernas skillnader mellan Covid-19 mRNA-batcher inkluderar:
- Kan oönskade händelsevariationer bero på kvalitativa eller kvantitativa varianser i mRNA-sekvenser eller antal mRNA-strängar mellan batcher?
- Kan oönskade händelsevariationer bero på kvalitativa eller kvantitativa variationer i storleken/morfologierna eller kvantiteten av LNP:er mellan batcher? Vilka tester har gjorts för att garantera säkerheten för olika LNP:er används i mRNA-injektioner?
- Var de partier som motsvarade de gula mot gröna mot blå datapunkterna på något sätt kvalitativt eller kvantitativt olika?
- Var lagring/hantering efter tillverkning äventyrad vid administreringsanläggningen (eller någon annanstans längs leveranskedjan) vilket ledde till produktvariabilitet?
- Vad är Sigma/felfrekvensen för denna och andra produkter som kommer från den specifika tillverkningsanläggningen/skiftchefen som ansvarar för tillverkningen?
- Var ingredienser från dessa av Covid mRNA-produkter hämtade från Indien eller Kina jämfört med någon annanstans, beroende på batch?
- Hur många procentandelar av partier av Covid-mRNA-produkter testades via personlig insamling av en FDA-inspektör jämfört med att ha "postats in" från början till idag? Testades varje enskild batch med endast någon av dessa två insamlingsmetoder?
- Utförde FDA verifiering av releasetest på de danska DKMA:s partier för rapportering av biverkningar? Om ja, varför släpper inte FDA dessa specifika testresultat? Om inte, varför gjordes inte testet?
- Finns det ett grundläggande problem med att konsekvent producera LNP:er och/eller mRNA-sekvenser på ett tillförlitligt sätt och utan kontaminering?
Resultaten av den danska studien och ovanstående frågor om biverkningar skulle kunna *börjas* behandlas, men inte utan att FDA självständigt delar med sig av resultaten av sina resultat från releasetesterna. Som det ser ut, på grund av allestädes närvarande FDA (b)(4) redaktioner, känner ingen till den validerade metoden för att testa Covid mRNA-skott or exakt vilka partier i den danska studien som testades eller inte or resultaten av dessa batch-tester.
…Återigen, även om FDA hade valt att släppa dessa batchtestresultat, hur vet konsumenterna om dessa resultat är representativa för specificerade batcher, eftersom tillverkarna själva väljer vilka prover som ska "postas in?"
Att inte tillhandahålla ingredienstransparens och säkerställa kvalitet via en lämplig provtagningsmetodik är ett grundläggande och grundläggande krav från FDA. I själva verket var det den främsta anledningen till bildandet av FDA! Förtjänar inte amerikaner bättre öppenhet, tillsyn och "sanningsfull märkning"-lagar när det kommer till våra läkemedel - särskilt eftersom dessa lagar stiftades för över 100 år sedan?
Publicerad under a Creative Commons Erkännande 4.0 Internationell licens
För omtryck, vänligen ställ tillbaka den kanoniska länken till originalet Brownstone Institute Artikel och författare.








